

Microtubules and Neurodevelopment Disease
Consisting of more than 86 billion neurons and 100 trillion synapses the human brain is arguably the most complex structure in the universe. Its construction depends on a complex cascade of biological processes that include mitotic division, relocation of migrating neurons, and the extension of dendritic and axonal processes. To gain insight into the molecules that mediate these cellular events the Keays laboratory has relied on unbiased phenotypic screens in mice, the generation of novel transgenic mouse mutants, coupled with the study of human neurodevelopmental disorders. This work has highlighted the importance of the microtubule cytoskeleton and microtubule associated proteins in the building of the human brain.
The Tubulins
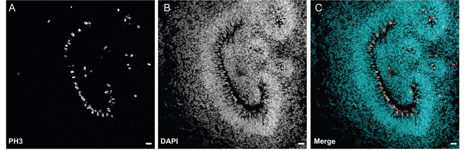
We have previously shown that mutations in the alpha tubulin gene TUBA1A cause lissencephaly a neurodevelopmental disease characterised by a smooth cortex, epilepsy and severe intellectual impairment (Keays et al, Cell 2007). The tubulins are a multi-gene family that encode for the constituents of microtubules; in humans there are 8 α- and 9 β-tubulin genes. Since our discovery that mutations in TUBA1A cause lissencephaly a host of neurological disease states have been associated with the tubulins. For instance, it is now known that variants in TUBB3 cause defects in axon guidance, mutations in TUBA4A cause motor neuron disease, and mutations in the β -tubulin gene TUBB5 cause microcephaly (Breuss et al, Cell Reports 2012). We are interested in understanding how mutations in the tubulin genes cause neurodevelopmental disease. To do so we are exploiting the power of human cerebral organoids to model the tubulinopathies. Drawing on our established network of clinicians, we generate iPSCs, which are then employed to generate “mini brains” (See Figure). We carefully analyse these organoids alongside controls that have been subject to genetic repair using the CRISP-cas9 genome editing system. We use a variety of methods including brain clearing (iDISCO), light sheet microscopy, live cell imaging, and transcriptomic methods to gain insight into the underlying molecular pathology.
The Masts
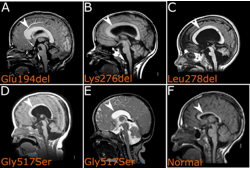
To understand the basis of neurodevelopmental disorders the Keays laboratory has undertaken genetic analysis of patient samples. This has revealed that mutations in an uncharacterised microtubule associated serine threonine kinase (MAST1) causes an unusual syndrome characterised by a “mega” corpus callosum and cortical malformations, as well as autism. Intriguingly, many of these mutations are single amino acid deletions in a domain of unknown function. There are four MAST family proteins (MAST1-4) that are largely uncharacterised and MAST3 has recently been implicated in Rett syndrome. Our goal is to define their expression pattern of the MASTs, identify their phosphorylation targets, understand how they regulate microtubule function, and explore how mutations cause neurodevelopmental disease. To achieve we have generated a variety of novel mouse models that recapitulate patient mutations, and reprogrammed iPSCs for organoid development.
Relevant Papers
Keays et al, Mutations in α-tubulin cause defects in neuronal migration in mice and lissencephaly in humans. Cell. 2007 Jan 12;128(1):45-57.
Gstrein et al. Mutations in Vps15 perturb neuronal migration in mice and are associated with neurodevelopmental disease in humans. Nature Neuroscience. (2018) Feb;21(2):207-217.
Tripathy, et al. Mutations in MAST1 cause mega corpus callosum syndrome and cortical malformations. Neuron. 2018 Dec 19;100(6):1354-1368.
Collaborators
Professor Jamel Chelly, Professor Nick Cowan (NYU), Professor Juergen Knoblich (IMBA, Vienna)